ChemXDyn: New framework to capture true chemistry behind complex reactive systems
The transition to net-zero and carbon-free fuels rests on a precise understanding of how molecules break apart and recombine during combustion and energy conversion. Predictive models of these processes, known as chemical kinetic models, are essential for designing cleaner engines, alternative fuels such as hydrogen and ammonia, and next-generation catalysts. Yet, constructing such models from experiments alone is difficult, as the underlying reaction mechanisms involve numerous elementary steps and short-lived intermediates that are seldom amenable to direct experimental characterisation.
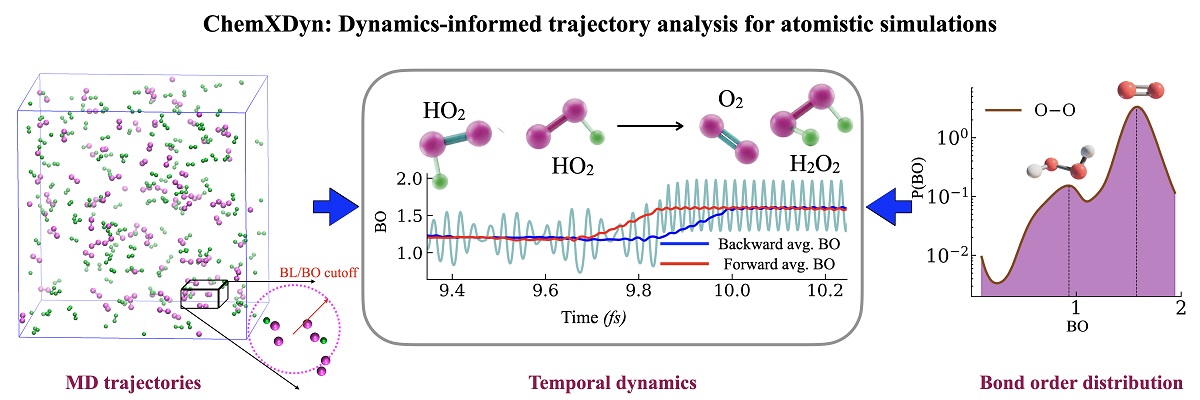
Molecular dynamics simulations offer a powerful complement by tracking the motion of every atom in a reacting system. Translating these atomistic trajectories into a useful chemical kinetic model has remained a long-standing challenge. Conventional analysis tools decide whether two atoms are bonded by comparing their distance to a fixed threshold at each instant. This simple criterion often mistakes brief, accidental encounters for genuine chemical bonds, generating spurious molecules and unreliable reaction rates. Existing dynamics-aware alternatives partially address this issue but rely on empirical, system-specific thresholds.
In a recent study led by Konduri Aditya and Phani Motamarri, along with their students Raj Maddipati and Dhruti Boddapati from the Department of Computational and Data Sciences (CDS), in collaboration with Elangannan Arunan from the Department of Inorganic and Physical Chemistry (IPC), the team introduced ChemXDyn, a computational framework that interprets simulation trajectories through the lens of time rather than instantaneous snapshots. By tracking how interatomic distances evolve and by enforcing fundamental rules of chemical bonding, ChemXDyn distinguishes true bond-forming and bond-breaking events from transient interactions.
The method was validated on the oxidation of hydrogen, ammonia, and methane, three reactions central to clean-energy technologies. ChemXDyn eliminated unphysical intermediates, recovered reaction pathways consistent with experiment, and produced more accurate reaction rates. The framework offers a reliable foundation for studying combustion, heterogeneous catalysis, plasma chemistry, and electrochemistry, supporting the discovery of sustainable energy solutions.
REFERENCE:
Maddipati R, Boddapati D, Arunan E, Motamarri P, Aditya K, ChemXDyn: Dynamics-Informed Species and Reaction Detection Methodology from Atomistic Simulations, Journal of Chemical Theory and Computation (2026).
https://pubs.acs.org/doi/10.1021/acs.jctc.6c00242
LAB WEBSITES:
https://cds.iisc.ac.in/faculty/konduriadi/
https://flamelab-iisc.github.io/
https://cds.iisc.ac.in/people/phani-motamarri/
https://ipc.iisc.ac.in/~ea/


